
1990年,人类基因组计划取得了决定性成功,我国也与1999年加入此计划。生物信息学是近些年迅速兴起的边缘学科,主要以计算机为工具对生物信息进行存储、检索和分析。DNA测序数据包含了极其重要的生物学信息,它所蕴含的生命科学仍然值得科学家进行深度挖掘。随着基因测序项目的展开,以及DNA测序技术成本越来越低和测序平台的不断普及,每天都在产生海量的DNA测序数据。如何解决这些庞大的数据量,已经成为当前学术界研究的热点话题,而最有效、最直接的方式便是数据压缩。
DNA测序数据与其他待压缩数据有所区别,DNA数据有其固定的特点,对其进行压缩时需要无损压缩。自从1993年Grumbach等提出针对DNA序列的压缩方法BioCompress,就意味着对DNA序列压缩的研究正式开始。在此背景下,越来越多的学者加大了对该领域的研究力度,提出了具有代表性的压缩方法,例如统计法、替代法等。在替代法方面,主要有Biocompress、CTW-LZ和DNACompress等。代表性的统计压缩算法包括CDNA和XM等。Giancarb等对传统DNA压缩算法做了综合性论述,针对于小规模的DNA测序数据来说,该压缩方法的效果更好。但是现阶段DNA测序数据呈现出高通量的特征,导致此方法的压缩效率偏低。
1.相关概念
1.1DNA序列
DNA属于高分子聚合物的一种,脱氧核苷酸是DNA的基本单位。每个脱氧核苷酸都是由一个碱基(Base)、一个磷酸分子(P)以及一个脱氧核糖(S)构成的。DNA分子中仅有4种碱基存在,它们分别是:腺嘌呤(A)、鸟嘌呤(G),胞嘧啶(C)、胸腺嘧啶(T)。脱氧核苷酸主要是通过化学键相互连接成线性排列成有方向的多聚核苷酸长链。这就是DNA的一级结构,可简单地按正方向顺序来书写的碱基序列来表示。
1.2高通量DNA测序相关的数据格式
在DNA测序技术快速发展的背景下,所形成的DNA序列数据越来越庞大,进而在存储、分析、管理以及传输方面面临着更多的问题。因此,出现了专业的数据格式,主要有SAM/BAM格式、FASTQ格式以及FASTA格式。FASTA格式数据通常由DNA序列的测序过后的拼接、组装等技术产生。SAM格式文件主要应用于测序序列的短读匹配到参考基因组上的结果。BAM格式文件是SAM格式文件的二进制形式,BAM格式文件的存储空间消耗更小。
1.3数据压缩基础
所谓数据压缩,可将其理解为在任何信息都不丢失的情况下,来对消息中的冗余进行去除,进而实现数据存储空间的减少,这对于实现存储、传输及处理效率的提升有着重要的意义。现阶段,主要可将数据压缩划分为无损压缩、有损压缩两个分支。其中,有损压缩的根本目标是促进压缩率的提升,进而可以存在部分精度的损失。无损数据压缩,则在一个压缩/还原周期之后,得到一个精确无差的原始输入数据流的副本。DNA序列数据蕴含着大量人类未知的生物学信息,因此只能够运用无损压缩技术来对数据压缩。其中,图1为数据压缩的一般步骤,其主要是由建模、编码两个阶段构成的。
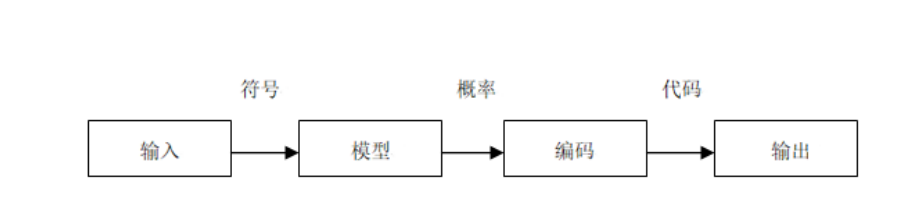
图1 数据压缩一般步骤
在无损压缩模型方面,是由字典模型、统计模型两个类别构成的。其中,统计模型的原理是按照顺序将字符读入,然后对字符出现的概率进行计算,在此基础上来分配变长码,最终达到压缩的目标,其中较为常用的方法有算术编码、Hufifiman编码等。而字典模型的原理则是在读入字符的过程中,运用字典将一连串的字符编码为一个标识,较为常用的技术有LZ77、LZ78等,其压缩效果与建模过程有着密切的关系。
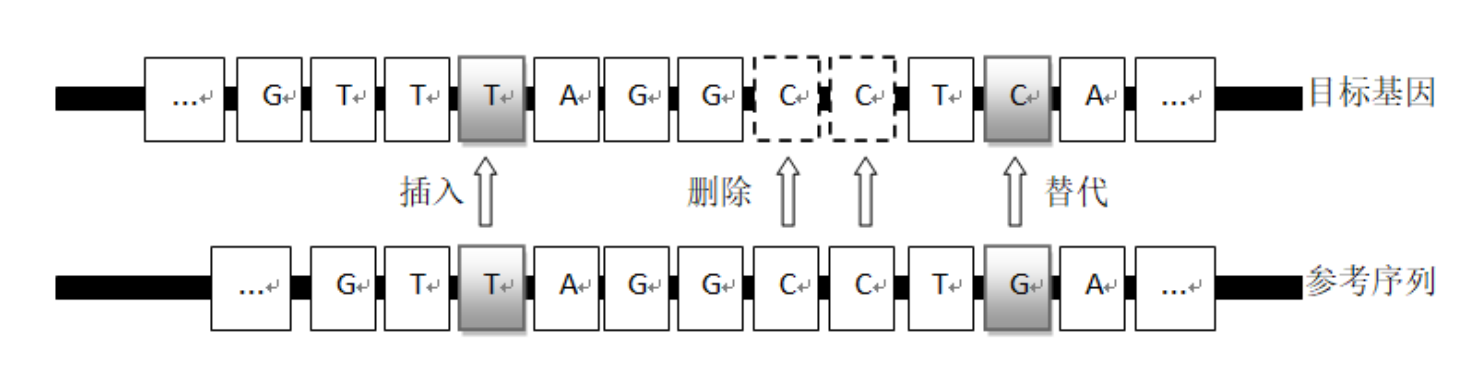
图 2 基于参考序列的DNA压缩技术原理图
(1)统计模型。在运用统计模型的过程中,第一步需要对可预测输入数据流中符号的概率进行考虑,然后按照得到的概率须脱离均匀分布。根据压缩环节是否需要进行更新,可将统计模型划分为自适应模型、静态模型两种。其中,在自适应模型中,编码器与解码器需采用相同的模型,同时无须从编码器传递统计表。静态模型则是指在压缩前,来对输入流进行扫描后形成统计表,在编码期间信源符号的概率是一直不变的。与静态模型相比,自适应模型能够更好地适应变化的数据,从而得到较高的压缩比。
(2)字典模型。字典模型是由自适应字典模型、静态字典模型构成的。其中,前者能够很好地处理各种类型的问题,因此当前所运用的字典模型基本上都属于自适应的;后者在压缩期间需与压缩后的数据共同存储,所占用的空间较大。
2.高通量DNA测序数据压缩方法
近年来,随着多国政府启动个人基因组计划的启动,基因测序技术逐渐普遍应用于各大领域,比如说精准医学、法医生物学、生物系统学,DNA序列已经成为不可或缺的知识。现阶段,高通量DNA测序方法中所运用到的平台主要有:AppliedBiosystemsSOLiD测序仪、Roche454测序仪、Illumina测序系统、PacBioRSII单分子测序和IonPGM以及Proton半导体测序仪。在DNA测序进行期间,得到可靠的数据是非常重要的,那么最直接、最有效的方法便是将短读对目标序列处于几十倍的覆盖率,意味着在此背景下所形成的数据量是相对庞大的。针对于下一代测序来说,根据短读组装、拼接模式的差异,具体可将其划分为从头测序、重测序两个类型,下面来对其做出详细的介绍。
2.1重测序DNA序列压缩
重测序主要是对已完成测序物种的不同个体而开展的测序。当前,因为受到人类单核酸多态研究、设计标记以及疾病研究等相关因素的影响,为了能够得到准确的结果,则需对同一物种的不同个体开展测序。当然,假如已经取得了该物种某个个体的完整基因组序列,则可运用下一代测序技术,将已知序列作为参考样本,进而形成大量DNA短读高度覆盖目标基因组,最后借助映射来得到目标基因组数据。
重测序数据压缩一般情况下也被称之为基于参考序列压缩,重点是根据记录参考序列和短读间的差异信息,来实现对数据与信息的压缩。众所周知,自然中同源物种基因组间的相似性是非常高的,因此重测序数据压缩所得到的压缩比是很高的水平。举例来说,任何两个人的基因组的相似性都超过了99%,假如已经获得了参考基因组,那么只需要对1%的差异信息进行存储即可。
2.2从头测序数据压缩
从头测序与重测序间存在着较大的差异,最明显的便是前者直接对待测个体开展测序,然后对形成的短读进行拼接、组装。现阶段,下一代测序短读拼接算法也是非常多的,其中较为常用的有OLC算法、DeBruijin图算法和基于OLC或DeBruijin图的贪婪算法。由于从头测序压缩与参考序列没有关联,因此有着较好的自完备性。但是需注意的是,拼接技术的好坏直接会影响到其压缩结果。总的来说,在已知参考序列的情况下,则采用重测序压缩方法的效果更好。
3.基于参考序列的生物DNA测序数据压缩技术
近些年,实施的基因组计划等相关项目,借助下一代测序技术形成了大量的DNA测序数据。而在DNA研究未来发展的进程中,对DNA测序数据的传输、存储的处理情况则处于重要的地位,那么则需要发挥出基于参考序列的DNA测序数据压缩技术。其中,图2为基于参考序列的DNA压缩技术原理。
在对压缩参考样本进行选择时,有必要选择合理的、恰当的参考序列。一般情况下,会选用拥有高度相似性的同源物种序列。然后,根据基于生物学特性的映射过程,来对短读与参考基因组的匹配位置、匹配类型、差异位置、差异类型、差异内容以及短读长度进行确定,其中,匹配位置是指距参考基因组起始点的位置。匹配类型是由4个类别构成的,1、2、3、4分别代表直接重复、镜像重复、配对重复、互补回文;差异位置是指相对短读首字符位置;差异类型由3个类别构成,1、2、3分别代表插入、删除、替换;差异内容中的1、2、3、4则分别对应A、G、C、T;短读长度则是指可变或固定长度。最后,需运用高效编码来对映射结果执行压缩。
总的来说,短读映射在基于参考的NDA数据压缩中处于核心地位,其根本目标是来对短读和参考序列间的差异信息进行寻找。现阶段,基于BTW的映射算法、基于哈希表的SFE算法是非常常见的。另外,在压缩映射结果方面,运用较多的方法有Huffffman、Gamma以及Delta等。一般情况下,差异位置、匹配位置会选择相对位置,这对于促进压缩性能的提升有着重要的意义。与此同时,由于DNA数据具有近似重复、精确重复的特征,这对于提升压缩率也有着积极作用。
伴随着基于参考序列的DNA数据压缩方法的增多,所取得的成果也越来越显著。现阶段运用最多的算法有GRS、BWB以及DNAzip等,主要原因是以上几种方法在压缩比方面有着明显的优势,但是面临着两个待解决的问题:其一,在解压缩的过程中,需要将参考序列作为参考指标,因此事先应将其存储在本地;其二,这些算法对参考基因数据有着非常强的依赖性,同时现阶段并不是所有的物种都有相关的参考序列。总的来说,在算法压缩数据使用期间,会受到参考序列的影响。
4.结语
在DNS测序数据不断发展的进程中,现阶段已经成为生物医学领域数量增加最快、应用最广的数据,对这些数据的管理、分析及以及应用给生物信息学带来了前所未有的挑战。自从DNA数据压缩技术提出以来,学术界也获得许多研究成果。在此背景下,所提出的序列压缩算法具有实用、高效等特点。需注意的是,即便此算法在实现DNA数据存储成本的降低方面取得了一定的成果,但是仍然在一些方面处于初步阶段,也引起了国内外学者的重视。总而言之,DNA测序技术给计算带来了极大的挑战,如数据存取、数据运算分析、数据应用和人才培养。想要实现生物医学领域的更好、更快发展,则当前亟需解决的问题便是DNA测序数据的传输、存储,意味着仍然需要设计更高效的DNA测序数据的压缩方法。
作者:彭棋
本文刊发于《中国高新科技》杂志2020年第22期
(转载请注明来源)





